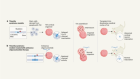
Big business
Biological drugs (biologics) — pharmaceutical drugs that are based on biological molecules such as proteins — accounted for almost one-quarter of drug spending worldwide in 2015. That fraction is set to continue to grow in the years ahead. Most biologics that achieve huge sales are targeted at common medical conditions. Drugs that tackle various types of cancer, diabetes and specific immune disorders represented more than half of global spending on biologics in 2015.

Sources: left, EvaluatePharma; right, IQVIA
A growing global market
Biosimilars are copycat versions of biologics that have typically reached the end of their patent protection. The European Union established a regulatory framework for biosimilars in 2003, and Japan followed in 2009. The United States created a regulatory pathway in 2010, but the first formally designated biosimilar hit the US market only in 2015. India set up formal guidelines for validating biosimilars in 2012, with refinement in 2016.

Sources: US, FDA; EU, EMA; India and Japan, GaBI Online
Sales impact
Many biologics that would otherwise experience a steady or growing demand show a sharp loss of sales on the launch of their biosimilars.
Avastin (bevacizumab; cancer) Biosimilar Mvasi has been approved for EU and US markets, but has reached neither.

Source: F. Hoffmann-La Roche
Herceptin (trastuzumab; cancer) Biosimilar Ontruzant hit the EU in 2018; no Herceptin copy is sold in the US market.

Source: F. Hoffmann-La Roche
Remicade (infliximab; immune disorders) Several biosimilars went on sale in the EU in 2015 and in the United States in 2016.

Sources: US, Johnson & Johnson; EU, Merck & Co.
Blockbusters on the brink
Time is running out for the manufacturers of some of the most lucrative biologics, with patents expiring in crucial markets over the next five years. Biosimilar developers are prepared, with pre-approved drugs ready to launch when these markets open. Notably, some biologics are covered by further patents related to their manufacture; these continue to offer protection after the original drug patent has expired.

Sources: top, AbbVie (Humira)/F. Hoffmann-La Roche (Herceptin, Avastin, Lucentis)/Regeneron (Eyelea)/Johnson & Johnson (Stelara)/Novartis (Lucentis); bottom: GaBI Online
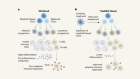
On track for approval
Biosimilars are more costly and difficult to produce than generic versions of small-molecule drugs. It can take 5–10 years and an investment of US$100 million–250 million to bring a biosimilar to market, compared with about 2 years and $1 million–10 million to develop a conventional generic — particularly in the complex patent landscape of the United States.

Credit: Mohamed Ashour/Andrew Khosravani
1. Reverse engineering The original biologic is analysed with methods such as mass spectrometry to reveal its amino-acid sequence, protein structure and any chemical modifications. These profiles will be compared with those of prospective biosimilars.
2. Cell-culture conditions Even when following the same genetic instructions, different cell lines can produce variants of a particular protein. Biosimilar developers must therefore identify an appropriate cellular factory, as well as optimize those cells’ growth conditions, to ensure that their product closely resembles the original biologic.
3. Testing the function Various assays are used to test how well a prospective biosimilar binds to its biological target, and to confirm that the drug replicates the effect and specificity of the original biologic.
4. Finding the formulation If a biologic is not properly prepared or mixed, it can misfold, degrade or aggregate. Consequently, biosimilar developers must identify manufacturing methods that result in a stable, reliable product.
5. Clinical confirmation Testing a biosimilar in people is faster than evaluating a biologic. Typically, only a phase I trial to show that the drug is safe and a phase III trial to show that it has an efficacy similar to that of the original are needed.
6. Regulatory review On the basis of the clinical data, a regulatory authority decides whether the a biosimilar is sufficiently similar to the original biologic. Further testing in people might be required.
 Nature Outline: Biosimilars
Nature Outline: Biosimilars
 Biosimilars: mimicking biological drugs
Biosimilars: mimicking biological drugs